PETIMOT:基于SE(3)-等变图网络的蛋白质运动推断框架
论文信息
| 字段 | 内容 |
|---|---|
| 标题 | PETIMOT: A Novel Framework for Inferring Protein Motions from Sparse Data Using SE(3) Equivariant Graph Neural Networks |
| 作者 | Valentin Lombard, Sergei Grudinin, Elodie Laine |
| 机构 | Sorbonne Université, CNRS; Univ. Grenoble Alpes, CNRS |
| 论文地址 | https://arxiv.org/abs/2504.02839 |
| 代码地址 | https://github.com/PhyloSofS-Team/PETIMOT |
| 发表时间 | 2025年3月(arXiv预印本) |
一句话概要
蛋白质的构象异质性对生物功能至关重要,但现有方法或依赖昂贵的仿真(分子动力学、流匹配),或局限于同源家族。论文提出PETIMOT框架,将问题重新定义为从PDB稀疏实验结构预测协方差矩阵的特征空间,利用SE(3)-等变图神经网络结合预训练蛋白质语言模型,设计对称感知损失函数(最小二乘、正弦子空间、独立子空间),在跨家族泛化、推理速度和精度上显著优于AlphaFlow、ESMFlow和正则模分析。
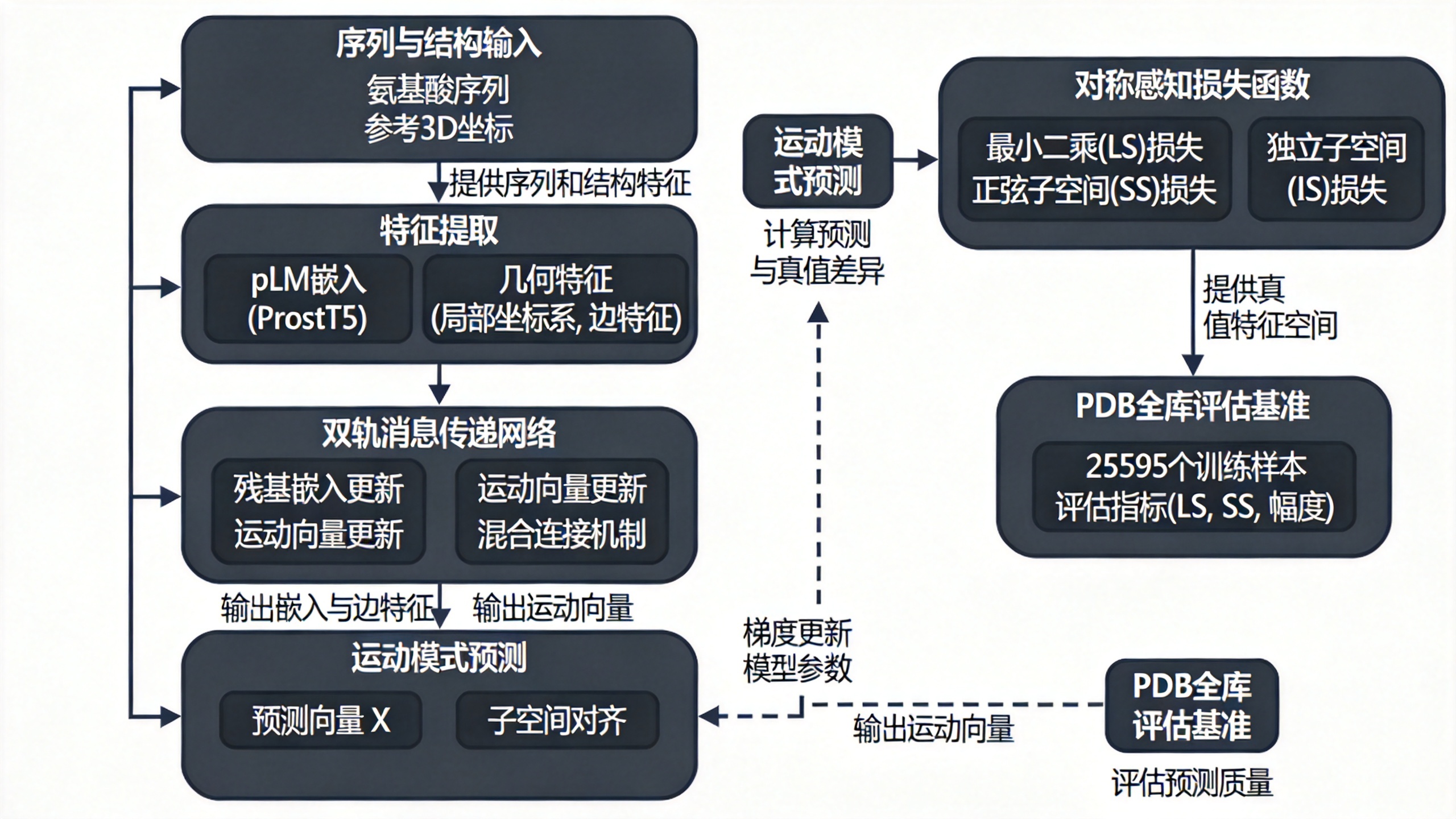
背景与研究动机
蛋白质通过构象变化执行功能,包括与配体、核酸、其他蛋白的相互作用。尽管AlphaFold2在单结构预测上取得突破,但模拟生理条件下的构象集合仍是基本开放问题。现有方法在预测替代构象、折叠开关、大幅度构象变化及溶液集合方面仍面临困难。
论文指出,同一蛋白或近源同源物的多个实验结构之间的多样性是溶液构象异质性的良好近似,且这种多样性通常可以由少量线性向量(模式)几乎完全解释。虽然线性空间不适合捕捉高度复杂的非线性运动(如环区变形),但具有学习更快、可解释性更强、推理更快、易于整合多数据维度等优点。
这一观察引导作者将问题重新定位:不是学习并从多维经验分布中采样,而是直接学习位置协方差矩阵的特征空间并跨同源层次泛化。
现有方法的瓶颈
-
单结构预测器的局限性:AlphaFold2等模型虽然精度高,但在生成构象异质性方面表现不佳。研究者尝试通过推理时干预(dropout、操纵MSA)产生多样性,但这些修改的效果难以解释和转移,尤其无法迁移到基于蛋白质语言模型的预测器。
-
基于扩散/流匹配方法的泛化不足:AlphaFlow/ESMFlow等虽然能生成高质量构象集合,但作者指出,跨家族训练的模型仍不能准确近似溶液集合,且容易产生幻觉。此外,这些框架需要生成大量样本(如50个),计算开销大——AlphaFlow在测试集上需38小时,ESMFlow需11小时。
-
家族特定模型的限制:基于变分自编码器或流形学习的方法(如DANCE、RAVE)能够在家族层面学习压缩表示,但无法推广到未见过的蛋白家族,限制了实际应用。
-
经典物理方法的精度瓶颈:正则模分析(NMA)计算效率高,但精度强烈依赖于初始拓扑,难以处理二级结构的大范围重排。
-
缺乏对称性感知损失:子空间比较任务需要损失函数对排列(不同模式的顺序)、尺度(振幅符号不固定)具有不变性,现有方法未针对这些对称性做显式设计。
核心洞察与贡献
论文的核心洞察是:与其从分布中采样构象,不如直接预测构象集合内在的低维线性运动空间。这相当于将问题从“生成”转化为“回归”——学习从蛋白质序列/结构特征到协方差矩阵特征空间的映射。由于特征空间是正交子空间,比较两个子空间需要特殊的几何损失,作者为此设计了三种具备对称不变性(排列不变、尺度不变)的损失函数。
主要贡献:
- 重新定义蛋白质构象多样性问题:将问题表述为从稀疏实验数据推断连续、紧凑的运动表示,而非从分布中采样。
- 构建基于PDB全库的结构多样性基准:使用鲁棒管道(DANCE)处理约75万条蛋白链,聚类后得到25595个训练样本,配备专用评估指标(最小LS误差、全局SS误差、幅度误差)。
- 设计SE(3)-等变图神经网络PETIMOT:采用双轨消息传递,同时更新残基嵌入和运动向量,融合来自预训练语言模型(ProstT5)的进化语义和局部几何信息。
- 提出三类对称感知几何损失:最小二乘(LS)损失、正弦子空间(SS)损失、独立子空间(IS)损失,均可组合,对排列和尺度具有理论不变性。
- 跨家族泛化能力:在824个测试蛋白上,PETIMOT的预测质量与训练集的结构/序列相似性无强相关,表明模型学到了可迁移的运动规则。
方法详解
数据表示与问题公式化
对于每个构象集合(同源蛋白链),先将序列对齐,利用权重wi(基于每个残基在集合中出现的比例)进行加权结构叠合。然后计算覆盖加权的Cα原子位置协方差矩阵:
其中R是3N×m的位置矩阵,R_0是参考构象坐标,W是3N×3N的对角覆盖矩阵。对C进行特征分解,得到特征向量矩阵Y(3N×K)作为真值运动模式。
模型的输入是蛋白序列和参考结构的3D坐标,目标是预测向量X ∈ R^(3N×L),使其张成的子空间与真值子空间Y在加权意义下对齐:
作者提出三种损失函数:
最小二乘(LS)损失:计算真值与预测向量对之间的加权最小二乘差异,通过线性分配问题(Hungarian算法)寻找最佳一一对应,并反传梯度仅通过匹配对。损失对全局缩放不变,因为优化缩放系数c_kl被解析求出。
正弦子空间(SS)损失:计算真值与预测子空间的正弦平方相似度。先将预测向量正交化(Gram-Schmidt),再计算所有对之间的均值:
该损失对子空间内的任意酉变换不变(包括正交化顺序),保证稳定训练。
独立子空间(IS)损失:替代正交化,通过加入预测向量之间的余弦相似度惩罚项(最大化子空间秩):
基线模型采用LS+SS等权重组合。
模型架构
PETIMOT采用消息传递神经网络,双轨更新:每个残基i维护一个嵌入向量s_i(来自ProstT5的维度d)和K个运动向量{x_ik}(在局部坐标系中,初始随机)。
对每个残基i,其邻居集合包括k个最近邻(按Cα距离)和l个随机选择的残基(k=5, l=10)。这种混合连接机制保证局部几何一致性和全局信息流,同时保持稀疏性,计算复杂度与序列长度N线性。
每个消息传递块中,邻居j的运动向量先通过相对旋转投影到i的局部参考系,消息由Message MLP处理(s_i, s_j, x_i, x_{ij}, e_{ij}),聚合后更新嵌入和运动向量。
局部参考系由每个残基的主链原子(N, CA, C)通过刚性变换T_i ∈ SE(3)定义,相对变换T_{ij} = T_i^{-1} \circ T_j对全局旋转平移不变。边特征包含:四元数编码的相对旋转、相对平移、序列分离度、空间距离的对数、以及所有主链原子对(N, CA, C, O)的径向基距离展开(16对×20基=320维)。
训练设置
使用AdamW优化器,学习率5e-4,权重衰减0.01,ReduceLROnPlateau调度(因子0.2,耐心10轮),早停耐心50轮。单张A100 GPU训练,每轮约9分钟。设置预测模式数K=4(与真值数一致),真值也取前4个主成分。
实验与结果
与基线方法对比
论文在824个测试蛋白上与AlphaFlow(OpenFold版,50个样本)、ESMFlow(50个样本)和正则模分析(NOLB,4个最低模)进行比较。实验设置中,对AlphaFlow/ESMFlow生成的构象集合提取主成分作为比对。
Table C.1. 成功率与平均性能对比
| 指标 | PETIMOT | AlphaFlow | ESMFlow | NMA |
|---|---|---|---|---|
| 成功率(LS<0.6)↑ | 43.57% | 31.80% | 26.82% | 24.88% |
| 推理总时间 ↓ | 15.82s | 38h 07min | 10h 41min | 43.59s |
| 最小LS误差 ↓ | 0.61±0.22 | 0.68±0.21 | 0.70±0.22 | 0.72±0.20 |
| 最小幅度误差 ↓ | 0.21±0.12 | 0.24±0.12 | 0.26±0.14 | 0.27±0.14 |
| 最优分配LS误差 ↓ | 0.83±0.10 | 0.86±0.10 | 0.87±0.10 | 0.88±0.10 |
| 全局SS误差 ↓ | 0.73±0.14 | 0.78±0.14 | 0.80±0.14 | 0.79±0.14 |
PETIMOT在所有指标上均取得最佳值,尤其成功率高约12个点,推理速度快2-3个数量级。值得注意的是,AlphaFlow和ESMFlow的训练数据可能包含部分测试蛋白(数据泄漏),而PETIMOT仍胜出。
批判性评估:baseline选择合理但不对称。AlphaFlow/ESMFlow是完整的构象生成器,PETIMOT只预测低维运动模式,两者的任务不完全相同。虽然论文通过提取生成集合的主成分进行公平对比,但要指出的是:流匹配模型能提供完整的构象分布(包括高强度运动和概率密度),而PETIMOT只提供K个线性方向,不提供运动幅度和分布性质。因此“精度更好”的结论应限制在线性子空间近似的语境中。
损失函数消融
对比LS、SS、IS单独使用与组合(LS+SS):组合模型在最小LS误差和全局SS误差上均优于任何单损失。IS单独使用表现最差,可能因为其缺乏对预测向量之间冗余的直接惩罚。SS损失在个体运动近似上偏弱,但子空间覆盖好;LS损失相反。组合平衡了两种需求。
序列与结构特征贡献
消融实验表明:去除结构信息(仅用pLM嵌入)后,ProstT5可部分补偿(因为其是结构感知语言模型),但ESM-Cambrian 600M表现骤降,说明结构嵌入是关键。用随机数替代pLM嵌入对性能影响很小,表明几何信息起主导作用,pLM嵌入提供边际增益。
图连接性消融显示:仅使用最近邻连接效果最差;引入随机连接(甚至完全随机连接)显著提升性能,说明全局信息流的重要性。
泛化能力分析
论文将测试蛋白的最大TM-score和序列相似性与预测误差(最小LS)绘制散点图,未观察到明显相关性。一些在训练集中无检测相似性的蛋白(TM-score<0.5)仍获得高质量预测,支持模型确实学到了跨家族的通用运动规则,而非记忆训练样本。
优势与局限性
优势:
- 任务重新定义带来的效率提升:预测低维子空间(4个向量)而非生成大量构象,推理速度比AlphaFlow快约8000倍,比ESMFlow快约2400倍。
- 对称感知损失函数的理论保障:LS、SS、IS损失均对排列和尺度具有不变性,特别是SS损失的酉不变性保证了训练稳定性和子空间覆盖质量。
- 结合pLM和几何信息的跨家族泛化:ProstT5的序列-结构双视图嵌入提供了比纯序列模型更强的适应性,消融证实几何信息主导。
- 可解释性:预测的运动向量可直接用于构象变形(如图3b-c展示的野生型经构象变化),便于可视化分析。
局限性:
- 线性假设的固有限制:线性子空间不能捕捉高度非线性的构象运动,如环区大幅度重排、折叠开关等。论文承认这一局限,但未提供量化的非线性误差分析。
- 只能预测固定K个模式:模型预测K=4个方向,真值也取前4个PCA分量。但不同蛋白的构象异质性维度不同,固定K可能丢失低频重要模式(如第5-8个PCA分量)。消融实验显示增加预测分量数量(1→4→8)可提升个体匹配能力,但8分量在最优分配指标上未显著优于4分量,说明4模式可能已覆盖主要方差,但该结论需更系统的验证。
- 仅使用Cα原子:忽略侧链和主链其他原子的运动,可能丢失与功能相关的精细构象变化。论文出于简化考虑,但实际应用可能需要多原子表示。
- 依赖PDB的采样偏差:真值PCA来自PDB中已有的实验结构数量,受数据库收录偏倚影响(如某些构象状态欠采样)。作者明确不预测特征值,因此无法区分方向的重要性。
- 对比方法的使用差异:AlphaFlow/ESMFlow需要MSA输入(AlphaFlow依赖OpenFold),而PETIMOT不需要,但PETIMOT需输入参考结构的3D坐标(对齐和计算边特征所需),且需要对应的结构集合进行训练。
可复现性评估:代码和数据已开源(GitHub),训练数据构建管道(DANCE)公开,超参数和训练设置详细。关键计算步骤(如下载PDB、聚类、特征提取)可复现,但依赖PDB-REDO等外部数据库,长期维护性不确定。
未来方向与开放问题
- 扩展到非线性运动:当前线性子空间假设限制了捕捉复杂构象变化。作者可引入核方法、流形学习或非线性模式(如自编码器)来增强表达能力,同时保持计算效率。
- 预测运动幅度:论文未预测特征值(运动方差),限制了重构真实集合的能力。联合预测方向与方差能输出更完整的构象概率信息。
- 整合多原子和侧链运动:扩展到全原子(甚至包括配体)可在药物设计等场景中提供更精细的动态信息。
- 序列到运动模式的端到端预测:当前模型需要参考结构输入;未来可探索仅从序列预测运动空间,直接结合pLM。
- 与其他生成模型融合:PETIMOT预测的运动方向可作为结构生成模型的先验或约束,引导扩散/流匹配在关键自由度上采样,减少幻象。
- 在动态蛋白质组学中的应用:将PETIMOT应用于大规模蛋白集合(如AlphaFold DB),预测所有蛋白的固有运动模式,构建蛋白质动态图谱。
组会预判问答
Q1: 为什么选择用线性子空间近似蛋白质运动?这在物理上合理吗?
- 论文依据:作者引述前人工作(Best et al., 2006; Lombard et al., 2024a),认为同一蛋白及其近源同源物的实验结构多样性可被少量线性向量解释。PCA/NMA在经典文献中已广泛用于描述蛋白质的主要构象变化(开放-关闭、铰链运动等)。论文指出线性空间在捕捉环区重排等非线性运动时有限,但选择了用线性模型换取计算效率和可解释性。
Q2: 与AlphaFlow/ESMFlow比较时,抽取生成集合的主成分再评估,这对流匹配方法公平吗?
- 论文回位:作者认为这种比较是合适的,因为流匹配模型输出的是构象集合,从中提取主成分与PETIMOT直接预测主成分属于同一任务——都是描述主要运动模式。但确实流匹配能提供更丰富的分布信息(如各构象的概率),而PETIMOT只给出方向不给出幅度。可以理解为任务本质不同:PETIMOT做回归(预测子空间),流匹配做生成(采样分布)。论文在比较时保持了方法中立,但结论“精度更高”需限定在子空间还原任务中。
Q3: 损失函数中对排列的线性分配使用scipy.optimize.linear_sum_assignment,这会导致梯度非平滑吗?作者如何处理?
- 论文原文:作者在文中明确表示已测试平滑版本的损失(连续梯度)但未改善性能。具体实现中,仅通过最优匹配对反传梯度,匹配选择本身不参与梯度计算(像“硬”对齐)。可以理解为这种做法在子空间比较任务中足够稳定,且实验显示性能良好。
Q4: 为什么训练时不需模拟数据,只靠PDB实验结构就能学到通用运动规则?这与蛋白质折叠物理有何关系?
- 论文依据:作者认为PDB中积累的天然状态实验结构(包括同一蛋白的多种晶体形式、系统发育同源物)蕴含了蛋白质生理条件下的主导运动信息,PCA能从这些数据中提取关键自由度。PETIMOT通过SE(3)-等变图网络学习结构特征与这些自由度之间的映射,并利用预训练语言模型提供的进化语义辅助泛化。这相当于从经验数据直接归纳运动规律,而非从头模拟物理力场。
本报告由立理AI生成,仅供参考,请以原文为准。